Introduction
inMOTIFin is a lightweight python package for simulating cis-regulatory elements. It consists of four command line modules and python access to the backend for advanced usage.
Simulation of sequences with inserted motif instances
This option of the inMOTIFin package is built using DagSim a simulation framework for causal models.
The directed acyclic graph (DAG) shown below describes the nodes and their relationship for the simulation. Each node describes a value or sampling strategy (depending on user input). Each node gets input from an upstream node (or the config file). Similarly, the output of each node is the input to its downstream nodes. This creates a dependency structure where each node is dependent only on the ones above it. For example: first groups are selected, then motifs from these groups, then instances from the motifs, then positions for each instance. As the motif-group membership information is used only when sampling motifs, the locations of the instances will not depend on the original groups the motifs were sampled from.
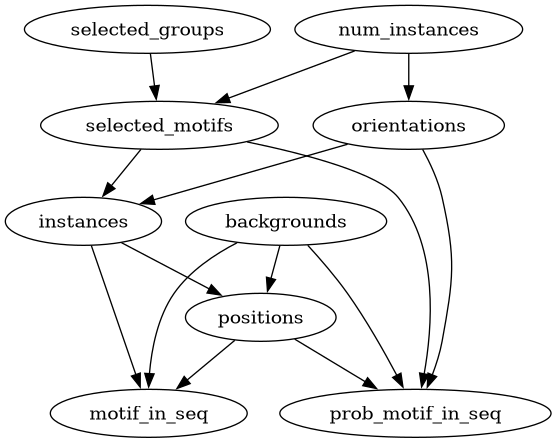
selected_groups |
Motifs are organized into groups. Groups have probabilities for being selected.
Within each group, motifs also have probabilities of being selected.
|
num_instances |
Number of motif instances per sequence. Options: integer or lambda parameter
for a Poisson distribution.
|
selected_motifs |
PWMs selected from a provided list (either simulated or from user input).
|
orientations |
The orientation of the motif instances is sampled from a binomial distribution.
|
instances |
List of instances sampled from the
selected_motifs. |
backgrounds |
List of backgrounds sampled from a provided list (either simulated or from
user input).
|
positions |
Positions of the inserted instances. Options: centering at the middle
of the background, aligning the right or left side of the instance to the
center, uniform sampling along the length of the background, sampling from
Gaussian distribution(s) from provided mean and variance values.
|
motif_in_seq |
Final sequence assembled from the sampled parts.
|
prob_motif_in_seq |
Position-base letter probabilities of the final sequence.
|
To read more about groups, visit the Explanation of groups page.
Simulation of random sequences
For i.i.d. simulation, the user can set minimum and maximum length, alphabet, and base preference that is shared across all sequences. When both minimum and maximum lenght is provided, the sequence length is sampled random uniformly between these two values (inclusive).
{'background_sim_seq_0': 'CGCGTACGGGGTCCGACCCTTAAAA',
'background_sim_seq_1': 'CCGCGAACGTGGCCGTCTGCAGAAC',
'background_sim_seq_2': 'AGAGCACCGGACGTGACCGCGGAAT',
'background_sim_seq_3': 'GTTCCACCCGAAACCACAGCGGCTA',
'background_sim_seq_4': 'ACCGTACAAAGGAGGTAAATCGATG'}
For context-dependent simulation, inMOTIFin can fit a hidden Markov model (HMM) on existing sequences and sample from the model. This requires a set of input sequences in FASTA format, expected alphabet (which may include more letters than the provided sequences, but not less), minimum and maximum length for sampling, and the order of the fitted model. When chosing an order, follow the considerations described by Charles E Grant in the MEME suite Google forum:
The larger the set of "background" sequences is, the better the results will be. (...)
The order of the Markov model is the number of preceding positions considered when calculating the character frequencies for the current position.
Typically, you should not specify an order larger than 3 for DNA sequences or larger than 2 for protein sequences. (...)
* For an accurate model of order N, you need to use a FASTA file as input (...) with at least 10 times 4**(N+1) DNA characters in it. So,
order-3 requires 2560 characters
order 4 requires 10240 characters
order 5 requires 40960 characters
etc.
Note that here the model is used to fit and sample from a HMM, so the motif-finding specific considerations are omitted (i.e. replaced with (…)). Also note that inMOTIFin can work with any alphabet, so it is not restricted to DNA or protein. However, it is important to note that the size of the alphabet has an impact on the model fitting capacity.
Simulation of motifs
by controlling their length, information content, and alphabet.
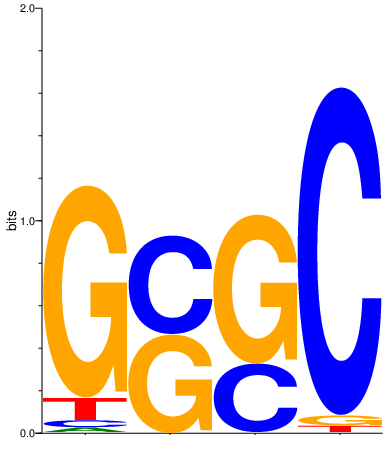

Motif_1 and motif_2 simulated from Dirichlet distribution defined with alpha values: A: 0.4, C: 1, G: 0.9, T: 0.5. For interpretation of values, visit the page motif simulation explained.
Multimerisation of motifs
by providing distances between the motif components.
Motif_1 and motif_2 multimerised by setting a positive distance.

…or a negative distance.
